
Proyectos de Investigación Genética
Nuestra investigación en el campo de la genética se centra en identificar factores de riesgo genéticos y en desarrollar nuevas dianas terapéuticas para el asma.
Resumen de proyectos
Nuestras poblaciones de estudio constituyen la base para nuestra investigación.
Saltar más abajo para ver:
- Ascendencia y mezcla
- Estudios de asociación genómicos (GWAS) y mapeo por mezcla
- Avanzando más allá de los GWAS: proyectos sobre factores de riesgo genético para el desarrollo de asma y fenotipos relacionados
- Metilación
Ascendencia y mezcla
Los latinoamericanos son descendientes principalmente de los amerindios pre-colombinos, africanos y europeos, mientras que los afroamericanos tienen proporciones variables de ascendencia africana y europea.
Los científicos han sido capaces de usar durante mucho tiempo marcadores genéticos para diferenciar poblaciones que han estado separadas geográficamente durante varias generaciones. Esto es posible porque la frecuencia relativa de los marcadores se diferencia entre grupos poblacionales que no se han mezclado con el paso del tiempo, un proceso denominado deriva genética.
Los avances recientes en genética nos han permitido avanzar más allá y poder estimar las proporciones de los grupos ancestrales en poblaciones mezcladas como los latinoamericanos o los afroamericanos.
Los conceptos de raza y grupo étnico son complejos, ya que incorporan características socioculturales y genéticas. Por esta razón, su uso en la práctica clínica es controvertido. Sin embargo, la raza y la etnicidad se usan para definir los valores normales en las pruebas de función pulmonar (PFTs) y se usan distintas ecuaciones para determinar los valores esperados de volumen pulmonar para afroamericanos, mexicanos y blancos. Dado que la función pulmonar medida se compara con el valor esperado calculado a partir de estas ecuaciones de referencia, asegurarnos de que esas ecuaciones son precisas tiene implicaciones importantes en el diagnóstico de enfermedad, en la determinación de la gravedad de la enfermad, en las reclamaciones de incapacidad y en la determinación del nivel de prioridad para los trasplantes de pulmón.
En el artículo “Ascendencia genética en la predicción de la función pulmonar,” publicado en Julio de 2010 en la revista New England Journal of Medicine, encontramos que las proporciones de ascendencia europea y africana en afroamericanos se asocian con la función pulmonar. La ascendencia genética se determinó usando datos genéticos de todo el genoma. También demostramos que si se incluyen estas medidas de ascendencia en la predicción de la función pulmonar, ésta mejora comparado con los estándares actuales clínicos que consideran a todos los afroamericanos como un solo grupo homogéneo.

Ahora estamos ampliando este trabajo mirando cómo la variación en ascendencia afecta la función pulmonar en otros grupos poblacionales mezclados, como los latinoamericanos. También estamos mirando otros rasgos, como los niveles de IgE, que se correlacionan también con la ascendencia genética. Finalmente, estamos extendiendo los conceptos de asociación con ascendencia con una técnica conocida como mapeo por mezcla. En ella, usamos programas informáticos sofisticados para inferir la ascendencia de cada región del genoma. Después miramos todo el genoma para ver si la ascendencia en cualquier región se asocia con un rasgo determinado o enfermedad. Nuestra hipótesis es que las variantes exclusivas o más comunes de una ascendencia se asocian con el rasgo o enfermedad analizados.

Estudios de asociación genómicos (GWAS) y mapeo por mezcla
Identificación de nuevos loci asociados con los niveles totales de IgE
La inmunoglobulina E (IgE) es un mediador clave de los procesos inflamatorios alérgicos y aparece frecuentemente elevada en las enfermedades alérgicas. Los niveles totales de IgE varían según la raza o grupo étnico en Estados Unidos, encontrándose mayores niveles de IgE en afroamericanos y latinoamericanos. Además, la ascendencia africana se ha asociado con mayores niveles de IgE en afroamericanos y latinoamericanos. A pesar de estas diferencias en niveles de IgE, sólo un 1% de las muestras analizadas en los GWAS anteriores de niveles de IgE fueron latinoamericanos.

Porcentaje de individuos incluidos en las fases de descubrimiento de GWAS previos por grupo poblacional.
En nuestros estudios identificamos nuevos loci asociados con los niveles de IgE en 3.334 niños latinoamericanos del Estudio Genes-ambiente & Mezcla en Latinoamericanos (Genes-environments & Admixture in Latino Americans study [GALA II]) empleando GWAS tradicional, mapeo por mezcla y secuenciación exónica de tres genes implicados en nuestros estudios. Replicamos las asociaciones encontradas en 454 latinoamericanos, 1.564 europeo-americanos y 3.187 afroamericanos procedentes de estudios independientes. Nuestros resultados resaltan la importancia de estudiar poblaciones multiétnicas diversas y el uso combinado de GWAS y mapeo por mezcla para descubrir nuevos loci asociados con los niveles totales de IgE.
GWAS y asma
Existen disparidades enormes en la prevalencia y mortalidad del asma entre distintos grupos raciales y étnicos, incluso entre aquellos grupos que normalmente se consideran uno sólo, como los mexicanos y los puertorriqueños. Gracias a que hemos recogido un gran número de muestras contamos con el mayor estudio de genes-ambiente en minorías étnicas de Estados Unidos. Esto permite a nuestro equipo de investigación identificar factores de riesgo genéticos, sociales y ambientales, los cuales ayudarán a explicar las grandes disparidades.
Más del 96% de los estudios actuales de asociación genética se han realizado en poblaciones europeas. Nosotros encontramos que más del 82% de los genes candidatos que se asocian con asma en otro grupos raciales/étnicos no se asocian con asma en mexicanos y puertorriqueños del estudio GALA. Entre los que se asocian con asma en este estudio, encontramos que algunos genes candidatos pueden considerarse “cosmopolitas” y son factores de riesgo tanto en puertorriqueños como en mexicanos. Sin embargo, otros genes son específicos de etnia y tienen efecto distinto en las dos poblaciones.

Además intentamos descubrir nuevos factores de riesgo para desarrollar asma en latinoamericanos y afroamericanos. Empleamos técnicas complementarias de estudios de asociación genómicos (GWAS) y mapeo por mezcla para identificar nuevas regiones del genoma que se asocian con asma.
En los GWAS, genotipamos simultáneamente cientos de miles de polimorfismos de un solo nucleótido (SNPs), variantes en el código genético que pueden conferir riesgo para desarrollar una enfermedad. Hemos verificado que una región del cromosoma 17, que se identificó originalmente en europeos, también se asocia con asma en latinoamericanos y afroamericanos.
En los mapeos por mezcla, usamos programas informáticos sofisticados para inferir la ascendencia de cada región del genoma, derivada de los ancestros africanos, amerindios o europeos, que son los tres grupos que se combinaron para dar lugar a los latinoamericanos actuales. Después miramos todo el genoma para ver si la ascendencia en cualquier región se asocia con asma. Con ello, hemos encontrado que tener ascendencia amerindia en la región del HLA, situada en el cromosoma 6 y que es importante para la función inmune, protege frente al asma en latinoamericanos. Tenemos proyectos en ejecución para secuenciar la región del HLA, que es una de las regiones más diversas del genoma, lo que nos permitirá identificar qué variantes y genes son responsables de la protección frente a asma.
Avanzando más allá de los GWAS
Secuenciación de genes candidatos
Los estudios de asociación genómicos (GWAS) han identificado numerosas asociaciones de variantes comunes con el asma y con fenotipos relacionados, incluyendo la respuesta a broncodilatador (BDR) y los niveles totales de IgE. En muchos casos las variantes identificadas por medio de los GWAS no son las mutaciones que causan enfermedad y, por tanto, debemos identificar estas variantes empleando secuenciación antes de realizar otros estudios.
Hemos secuenciado los exones codificantes más las regiones UTRs (regiones no traducidas, del inglés “Untranslated Regions”) de siete genes implicados a través de GWAS y mapeo por mezcla en 2.000 latinoamericanos, incluyendo un gen de susceptibilidad a asma (SMAD2), tres genes de BDR (SLC24A2, SLC24A4 y SLC22A15) y tres relacionados con los niveles totales de IgE (HLA-DQB1, HLA-DRB5 y ZNF365). Hemos identificado nueva variación genética asociada con respuesta a broncodilatador y con IgE dentro de tres de los genes secuenciados, incluyendo una contribución significativa de variantes raras no sinónimas en SLC24A2, SLC24A4 y HLA-DQB1. Nuestros resultados sugieren que la secuenciación masiva de genes identificados mediante GWAS y mapeo por mezcla es esencial para el mapeo fino de las asociaciones genéticas. Re-secuenciación en muestras mayores de poblaciones multi-étnicas y estudios funcionales se requieren para entender mejor el papel de las variantes raras y comunes en el asma y los fenotipos relacionados.
SMAD2 y asma
El gen SMAD2 (del inglés “Mothers against decapentaplegic homolog 2”) es una proteína implicada en la señalización del TFG-beta. En nuestros estudios en curso, realizados como parte del consorcio EVE, hemos encontrado que la ascendencia amerindia en SMAD2 se asocia con un mayor riesgo de asma en latinoamericanos. Este es un fuerte indicativo de que podrían existir mutaciones dentro o cerca del gen SMAD2 que contribuyan al desarrollo de asma en niños latinoamericanos.
Actualmente estamos secuenciando y haciendo estudio de la expresión génica para identificar la variación causal que subyace la asociación de la ascendencia amerindia en SMAD2 con el desarrollo de asma. A través del servicio de genotipado y secuenciación (RS&G) del NHLBI, estamos secuenciado el gen SMAD2 y el gen flanqueante (ZBTB7C), incluyendo la región no codificante que los separa, en 2.000 latinoamericanos con y sin asma. Nuestra hipótesis es que el locus SMAD2 contiene variantes raras específicas de población que contribuyen al desarrollo del asma. Además creemos que las variantes raras cercanas a SMAD2 también se asocian con distinto BDR y riesgo a presentar exacerbaciones del asma al modificar la expresión de SMAD2.
Secuenciación de exomas en extremos de respuesta a medicación
El albuterol es un β2-agonista de acción corta que es la medicación más frecuentemente prescita para el asma en todo el mundo. Existen diferencias marcadas en la respuesta terapéutica al albuterol entre distintos pacientes y grupos raciales/étnicos. Por ejemplo, los niños puertorriqueños y afroamericanos con asma responden peor al albuterol que los niños caucásicos y mexicanos. Por ello, estamos llevando a cabo estudios para entender las bases genéticas de la distinta respuesta a la medicación en niños pertenecientes a minorías étnicas con asma.
Nuestra hipótesis es que variantes raras, con efectos potencialmente mayores que los SNPs comunes, contribuyen a las diferencias raciales/étnicas en la respuesta a broncodilatador. Por ejemplo, en nuestros estudios de GWAS de respuesta a broncodilatador hemos identificado un exceso de p-valores de asociación muy significativos que se deben completamente a las variantes de baja frecuencia. Por ello, estamos explorando esta hipótesis en detalle realizando secuenciación de exomas y regiones UTRs en 1.500 afroamericanos, puertorriqueños y mexicanos. Todos ellos son pacientes con asma escogidos por presentar valores extremos de respuesta a broncodilatador. En concreto, estamos identificando variantes individuales o en conjunto que se asocien con BDR al comparar aquellos individuos que responden muy bien al broncodilatador frente a los que tienen muy poca respuesta. Además, en nuestros estudios en curso estamos evaluando la interacción de factores genéticos con factores ambientales que modifican la respuesta a broncodilatador.

Q-Qplot de un GWAS de BDR en GALA II. Los círculos negros indican todos los SNPs, los círculos azules muestran una disminución de la señal tras excluir las variantes menos comunes (frecuencia del alelo menor, MAF<5%).
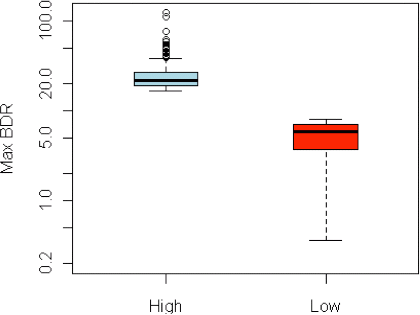
Valores máximos de BDR en individuos que responden muy bien al broncodilatador frente a los que tienen muy poca respuesta en 500 puertorriqueños, 500 mexicanos y 500 afroamericos de los estudios GALA y SAGE. Nota: los valores de Max BDR se muestran en una escala logarítmica.
La secuenciación de genomas completo ofrece la oportunidad de tener una visión más completa de las variantes genéticas que afectan a los caracteres complejos y a las enfermedades. Como parte del Consorcio de Asma en Poblaciones de Ascendencia Africana (CAAPA), estamos examinando los patrones de variación de los genomas de 500 casos con asma y 500 controles. Uno de los mayores objetivos de CAAPA es usar estos datos para desarrollar una plataforma comercial de genotipado que esté mejor diseñada para capturar el mayor grado de variación genética presente en los individuos con ascendencia africana.
Consorcio de Asma en Poblaciones de Ascendencia Africana (CAAPA).
Estudios del HLA
La variación genética del complejo mayor de histocompatibilidad (MHC) tiene un papel fundamental en la susceptibilidad al asma y en la regulación de los niveles de IgE. En nuestros estudios, la realización de mapeos por mezcla de asma e IgE en latinoamericanos indicaron que la variación genética en el MHC tiene una contribución significativa en ambos fenotipos. Específicamente, encontramos que la ascendencia amerindia en el MHC de tipo I se asocia con menor riesgo de desarrollar asma y que en el MHC de tipo I y II se asocia con menores niveles de IgE.
Actualmente estamos realizando estudios piloto de secuenciación de 5 Mb de la región MHC en niños latinoamericanos usando secuenciación de nueva generación de alta cobertura dirigida a esta región. Estamos integrando una tecnología avanzada de captura de la región MHC con métodos clásicos de caracterización del antígeno leucocitario humano (HLA). Usaremos estos datos para identificar los tipos de HLA y la variación genética de la región del MHC que se asocian con asma y niveles totales de IgE. Además, esperamos desarrollar un protocolo analítico que permita promover el estudio de la región MHC en latinoamericanos mediante la integración de datos de secuenciación de nueva generación dirigida, imputación de tipos de HLA empleando SNPs y métodos clásicos de caracterización del HLA.
Metilación
Existe una creencia de que el genoma prácticamente no cambia y que el código genético de una persona permanece intacto. Sin embargo, muchos procesos biológicos son capaces de encender y apagar la expresión de ciertos genes mediante lo que los científicos denominan epigenética (epi- es un prefijo griego que, en este caso, significa “por encima de”). Uno de esos procesos es la metilación, por la cual un grupo metilo se añade a ciertos patrones de pares de bases conocidos como sitios CpG, pero no cambian el código de ADN por sí mismo. La metilación cambia la probabilidad de que un gen se convierta a proteína. Nosotros hemos usado una tecnología avanzada de microarrays para obtener datos de metilación repartido por todo el genoma, correspondientes a cientos de miles de sitios CpG, y buscamos correlacionar estos cambios epigenéticos con el asma y fenotipos relacionados.
Tanto los factores genéticos como los ambientales son capaces de producir cambios en los patrones de metilación, si bien el alcance de cada uno de ellos no se ha establecido totalmente. Nosotros hacemos uso de la gran variedad y riqueza de datos genéticos y ambientales entre los participantes del estudio GALA II para explorar cuánto influyen los componentes genéticos y ambientales en los patrones de metilación. Hemos observado que hay diferencias en los patrones de metilación de distintos grupos étnicos, incluso entre grupos relacionados como puertorriqueños y mexicanos. Además, hemos visto que muchas de estas diferencias se deben a la distinta ascendencia de estos dos grupos. Estas diferencias en la composición ancestral se asocian con diferencias en los patrones genéticos, los cuales conducen a cambios en los patrones de metilación. Esto tiene un impacto profundo en las enfermedades complejas como el asma.