On this page
- Hit identification (Screening)
- High-throughput screening (HTS)
- Compound libraries
- Fragment discovery
- Tethering
- Hits-to-leads medicinal chemistry
Hit identification (Screening)
Small molecule hits are identified using a variety of experimental screening technologies, including:
- biochemical screens measuring a specific functional activity
- biophysical screens measuring a specific binding activity
- in vitro cell-based assays using cell line, primary cells and 3D mixed cell cultures or organoids
- in vivo small model organism screens using high-content imaging (e.g. Zebrafish or C. elegans)
- fragment screening by fragment screening by surface plasmon resonanceexternal site (SPR) and nuclear magnetic resonanceexternal site (NMR)
- site-directed disulfide trapping (aka tethering)
- covalent fragment or drug-like compound screening by mass spectrometry (MS)
See case studies of SMDC projects in Programs & Projects.
High-throughput screening (HTS)
High-throughput screening (HTS) uses miniaturized assays in high-density microplates (384- or 1536-well formats) with automation and robotic automation to allow testing of thousands to millions of compounds against one assay. Assays are miniaturized into 1-100 µl volumes and arrayed in plates containing ninety-six 1536-wells. The SMDC routinely uses 384-well format to test up to 400,000 compounds. Biochemical assay formats include measurements of fluorescence polarization, energy transfer, absorbance, luminescence, either as end-point or kinetic readouts. Cell-based assays can be based on whole well averages of a colorimetric, fluorimetric, or luminescent signal, or on high-content imaging of individual cells (bright field or fluorescence).
The SMDC has a recharge policy to facilitate development and executions of high-throughput screens. HTS projects tend to be highly collaborative and involve intellectual engagement from both the initiating labs and the SMDC staff scientists. Thus, contributions generally warrant co-authorship and staff often support grant submissions and fundraising efforts. Please do not hesitate to contact us with any questions and for screening costs. More details: High-throughput Screening Steps.
Compound Libraries
The SMDC houses more than 500,000 molecules, including libraries based on diverse compounds, target class-focused compounds, approved drugs, and bioactives with known mechanisms of action (see Available Compounds). Libraries conform to the current standards for lead-likeness. For the diversity collection, molecules were selected to have at least four lead-like properties (MW < 350 Da; cLogP < 5; H-bond donors < 5; H-bond acceptors < 8; rotatable bonds < 8; polar surface area < 100A2). Compounds were also filtered to remove reactive functionalities (e.g., Michael acceptors and thiols) and known toxicophores (e.g., nitroxides), and promiscuous scaffolds have been flagged [e.g., 6,305 compounds contain chemotypes flagged as pan-assay interference compounds (PAINS) (Baell, 2010)]. More than 100 screens have been run with most of the compounds in this collection, providing a rich data set for identifying artifacts and potentially mechanisms of action. Our web interface SMDC HiTS also links to compounds found in ZINC and PubChem ChEMBL, enabling us to access data for hundreds of additional screens.
Fragment discovery
Fragment-based drug discovery (FBDD) is a complementary technology to high-throughput screening (HTS) and has proven successful for many drug targets that are recalcitrant to traditional functional assays. By this approach, fragments—compounds approximately one-half the molecular weight of a drug—are assayed using biophysical or structural approaches. Fragments shown to bind to the target are then structurally characterized and are optimized through linking or merging two fragments, or via addition of designed functional groups to a single fragment.

Fragment screening is a special expertise of the SMDC. Routinely available technologies include surface plasmon resonance (SPR) and mass spectrometry (MS). Nuclear magnetic spectroscopy (NMR), and X-ray crystallography-based screens are also possible. The current state-of-the-art in fragment optimization is structure-guided design, and therefore we recommend developing a path to high-resolution structural information in concert with fragment screening.
Cys-tethering
Tethering is a site-directed fragment discovery method wherein bespoke libraries of disulfide-containing compounds are screened for binding to a native or engineered cysteine residue. Fragments that are complementary to the binding site and have the proper geometry to react with the cysteine residue are stabilized. Mass spectrometry or functional assays are used to identify bound fragments. The tethering approach was first developed at Sunesis Pharmaceuticals. The SMDC continues to develop this unique approach, inventing new synthetic methods for library production, and optimizing LC/MS-based detection methods for disulfide screening.
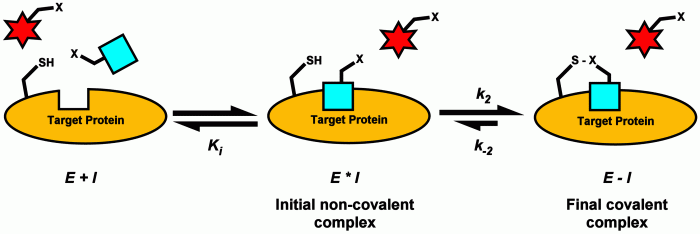
Hits-to-leads (H2L) medicinal chemistry

After a hit series is selected, we can move to initiate the purchase or synthesis of structural analogs. In general, the number of scaffolds taken forward into hits- to-leads medicinal chemistry depends on their attractive properties and the resources available to acquire and synthesize analogs. As a rule of thumb, each scaffold requires a half-time to full-time chemist during the early stages. We build a family of structurally similar compounds around the hits using several strategies. The primary data may suggest portions of the scaffold where structural changes modify function. Additionally, compounds are likely to have some sites that are easier to modify than others. The easiest way to scout for prospective structure-activity relationships (SAR) is to take advantage of commercially available materials. Web resources determine the availability of related compounds and allow searching based on substructure/scaffold or on overall similarity (e.g., Tanimoto similarity external site). Once materials have been received, quality control must be undertaken to assure the compounds have the correct mass and purity >95%. Its structure may also be confirmed by proton NMR. Pure compounds are then evaluated in the primary assay and key confirmatory, selectivity, and secondary assays. Ideally, this SAR-by-catalog exercise will show that small changes to the compound structure alter the potency without changing the mechanism of action. Eventually, commercially available analog aquisition will be exhausted, and compounds must be synthesized to address specific questions about the influence of certain molecular aspects or electronic, redox, or steric factors underlying the potency, selectivity, and pharmacologic activity.